Easy to Use
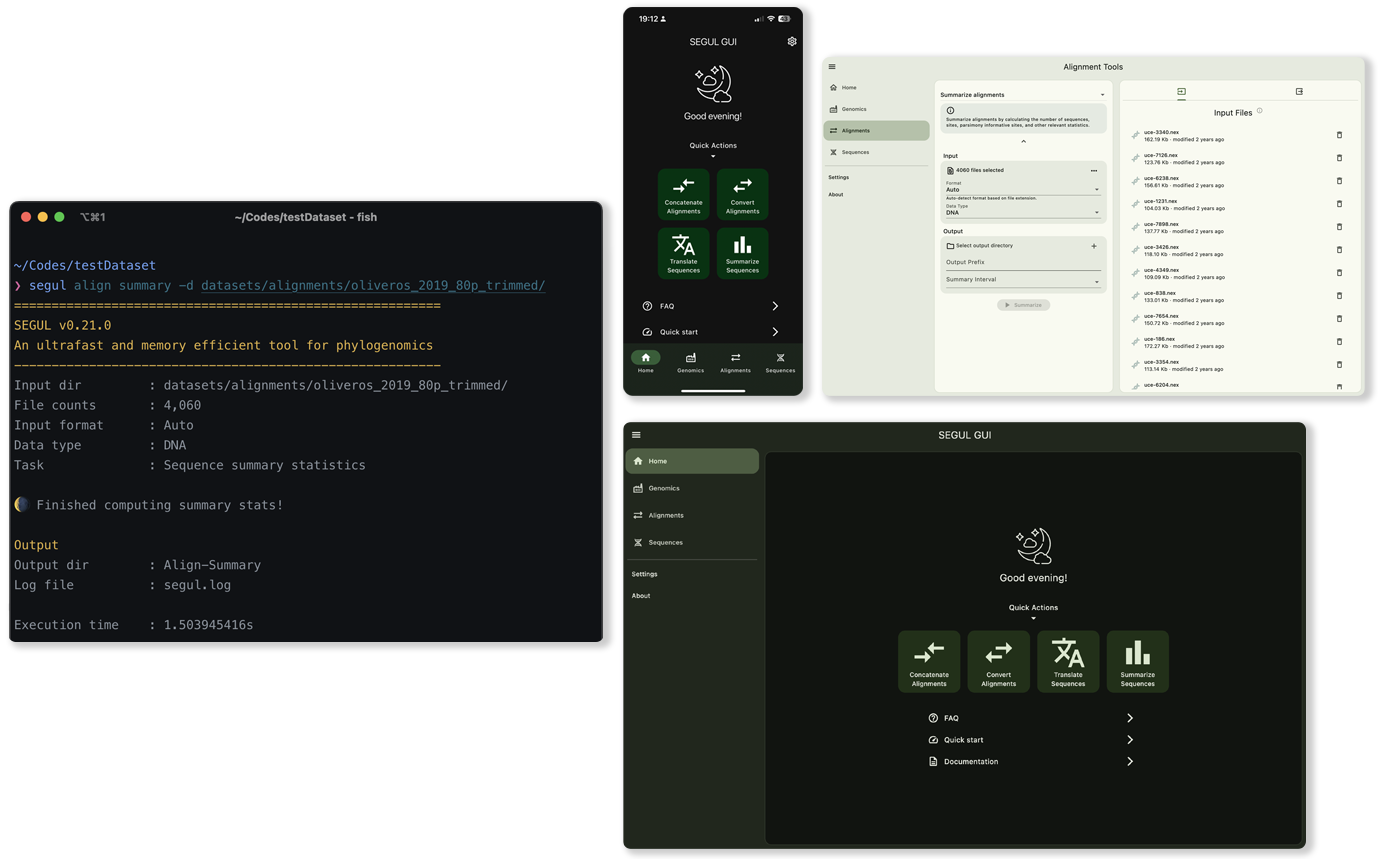
SEGUL offers an intuitive command line interface for efficient operation, along with a user-friendly, interactive graphical interface. We provide comprehensive documentation to help users with varying levels of technical expertise.
Accessible and Reproducible
SEGUL supports a wide range of devices, from smartphones to high-performance computers, without requiring any runtime dependencies. It also negates the need for supplementary applications like Docker or Singularity, enhancing its user-friendliness and accessibility.
Fast and Memory Efficient
SEGUL delivers high-speed performance while maintaining minimal memory usage. It’s engineered to leverage multi-core CPUs for rapid data processing, all without requiring manual intervention from the users.
Features
SEGUL features operation on alignment, genomic-specific, and sequence datasets.
Alignments
Concatenate, convert, filter, split, and summarize alignment datasets. Also convert between different partition formats. Support Sanger and next-generation sequencing data.
Genomics
Summarize FASTQ read and contiguous sequence datasets. Work on multiple large datasets simultaneously.
Sequences
Extract, map, rename, remove, and translate sequence datasets. Support Sanger and next-generation sequencing data.


What's New in SEGUL v0.23.0
New Features 🚀
- Convert alignments to unaligned sequences
- Add sequence to an existing sequence files/alignments
- Trim alignments
- New options for filtering alignments (see details):
- Minimum or maximum sequence length
- Minimum or maximum of parsimony informative sites
- Minimum taxon counts
- Based on user-defined list of sequence IDs
Breaking Changes ⚠️
- Filtering argument changes for better consistency (see docs)
- Remove
--ntaxoption from alignment filtering because by default SEGUL automatically and fastly counts the number of unique taxa across all input alignments
Bug Fixes 🐛
- Fix max-gap filtering issues
- Fix concatenation leading to missing data when sequence IDs in FASTA files contain trailing whitespace
Other Changes 🛠
- Migrate to Rust 2024 edition
- Update dependencies